
FDA beginnt, Tierversuche für Arzneimittelsicherheit mit KI-Modellen und menschlichen Organoiden schrittweise abzubauen
Eine stille Revolution in der Medikamentenprüfung: FDA-Bruch mit Tiermodellen verspricht eine schnellere, billigere und ethischere Biopharma-Pipeline
Die US-amerikanische Food and Drug Administration (FDA) hat heute angekündigt, die obligatorischen Tierversuche für monoklonale Antikörper und andere Medikamente schrittweise abzuschaffen. Die Entscheidung, die jahrelang vorbereitet wurde und von ethischen Verfechtern und klinischen Wissenschaftlern gleichermaßen gefordert wurde, setzt einen dramatischen Übergang zu besser vorhersagbaren, effizienteren und menschenrelevanten Methoden zur Bewertung der Arzneimittelsicherheit und -wirksamkeit in Gang.

„Es ist die Auflösung einer hundertjährigen wissenschaftlichen Annahme“, sagte ein mit dem Prozess vertrauter Analyst. „Wir bewegen uns weg von Tier-Modellen und hin zum Bereich der computergestützten Biologie und entwickelten menschlichen Systeme. Die Auswirkungen sind gewaltig.“
Vom Tierlabor zum Siliziumchip: Die bahnbrechende Roadmap der FDA
Eine neue Ära der Toxikologie
Die neu vorgestellte Roadmap der FDA umreißt die Verwendung von New Approach Methodologies (NAMs) – einer Reihe von Werkzeugen, die von KI-gestützten Rechenmodellen bis hin zu von Menschen gewonnenen Organoiden und Organ-on-a-Chip-Technologien reichen. Diese Werkzeuge zielen darauf ab, die menschliche Biologie mit weitaus größerer Genauigkeit als der derzeitige Standard – Tierversuche – nachzubilden.
New Approach Methodologies (NAMs) bezeichnen eine Vielzahl moderner Techniken, die hauptsächlich in der Toxikologie und Sicherheitsbewertung eingesetzt werden. Sie umfassen In-vitro-, In-silico- und andere nicht-tierische Methoden, die als Alternativen entwickelt wurden, um traditionelle Tierversuchsverfahren zu ersetzen, zu reduzieren oder zu verfeinern.
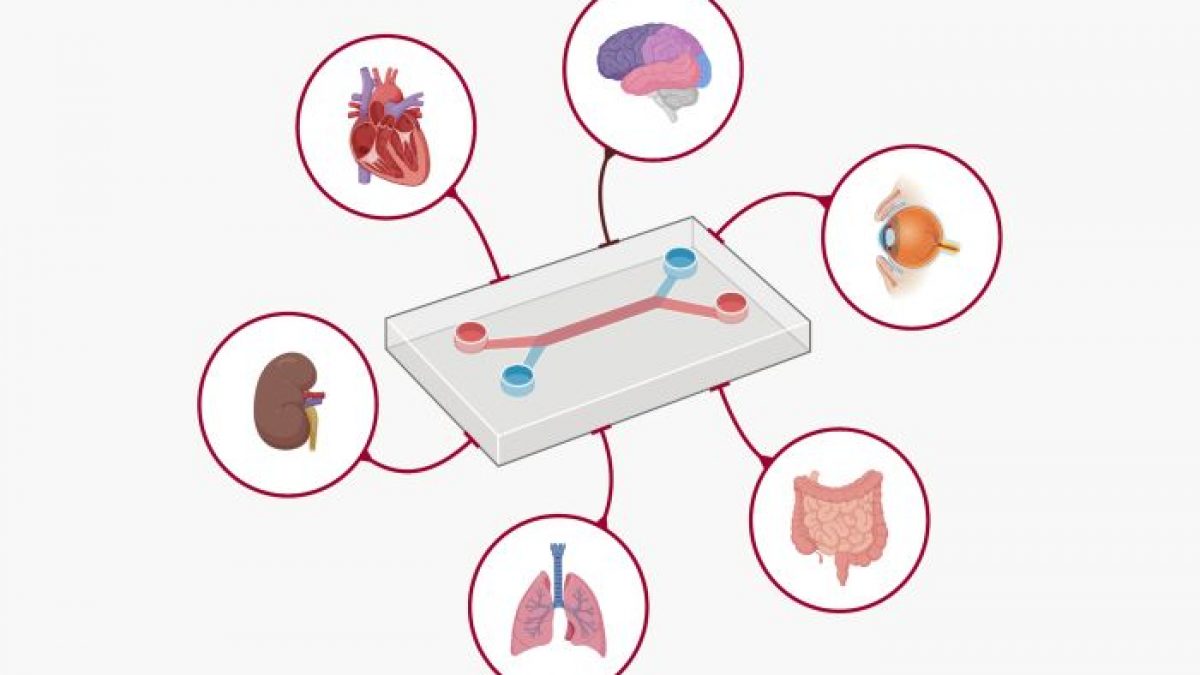
Zu den wichtigsten Bestandteilen der Richtlinie gehören:
- Sofortige Förderung von NAMs-Daten in Anträgen für neue Arzneimittelstudien (IND).
- Akzeptanz von Sicherheitsdaten aus der realen Welt von vergleichbaren internationalen Aufsichtsbehörden.
- Pilotprogramme, die in diesem Jahr beginnen und ausgewählten Entwicklern die Möglichkeit geben, monoklonale Antikörper einzureichen, die hauptsächlich mit nicht-tierischen Methoden bewertet wurden.
- Anreize, einschließlich beschleunigter Prüfungen, für Entwickler, die validierte nicht-tierische Testdaten vorlegen.
„Das ist eine Win-Win-Situation“, sagte ein leitender Toxikologe. „Medikamente kommen schneller zu den Patienten. Die Entwicklungskosten sind niedriger. Und wir entfernen uns von ethisch fragwürdigen, oft wenig aussagekräftigen Tiermodellen.“
Der Status Quo: Fehlerhaft, teuer und ethisch fragwürdig
Die versteckten Kosten von Tierversuchen
Seit Jahrzehnten sind Tierversuche der Goldstandard der Regulierung. Von Mäusen und Ratten bis hin zu Hunden und Primaten wurden Tiere verwendet, um menschliche Reaktionen auf Prüfpräparate zu simulieren. Aber diese Modelle werden oft nicht gerecht. Ein Molekül, das bei Nagetieren gut funktioniert, kann sich beim Menschen unvorhersehbar verhalten – eine Übersetzungslücke, die zu unzähligen gescheiterten klinischen Studien im Spätstadium und Sicherheitsentfernungen nach der Zulassung beigetragen hat.

| Kennzahl | Statistik/Erkenntnis | Quelle/Kontext |
|---|---|---|
| Gesamt-Fehlerrate klinischer Studien (nach präklinischen Tests) | Ungefähr 90 % bis 92 % der Arzneimittelkandidaten, die in klinische Studien am Menschen eintreten, erhalten keine behördliche Zulassung. | Mehrere Quellen zitieren diese Spanne, darunter BIO, Cruelty Free International und akademische Arbeiten, die sich auf Branchen-Daten beziehen (z. B. Sun et al., 2022). |
| Hauptgründe für das Scheitern in klinischen Studien | Mangelnde klinische Wirksamkeit (40-50 %), unüberschaubare Toxizitäts-/Sicherheitsprobleme (ca. 30 %), schlechte arzneimittelähnliche Eigenschaften (10-15 %). | Analyse zitiert in Sun et al. (2022) und bestätigt durch die Feststellung, dass über die Hälfte der Phase-II/III-Fehler auf mangelnde Wirksamkeit zurückzuführen sind. |
| Vorhersagbarkeit von Tiermodellen | Hohe Fehlerraten beim Menschen (z. B. 92 %) treten trotz vielversprechender Ergebnisse in präklinischen Tests, einschließlich Tierversuchen, auf. | Cruelty Free International, PETA, Humane Society International und akademische Überprüfungen heben die Diskrepanz (Übersetzungslücke) hervor. |
| Erfolgsrate nach klinischer Phase (ungefähre Daten von 2011-2020) | Phase I: ~52 % Erfolg; Phase II: ~29 % Erfolg; Phase III: ~58 % Erfolg. | Von BIO (Biotechnology Innovation Organisation) gemeldete Daten deuten darauf hin, dass Phase II ein wichtiger Engpass ist. |
| Fehlerrate nach Therapiebereich (ungefähre Daten von 2011-2020, Eintritt in Studien) | Urologie (~96 % Fehler), Kardiologie (~95 % Fehler), Onkologie (~95 % Fehler), Neurologie (~94 % Fehler). | Von BIO gemeldete Daten zeigen sehr hohe Fehlerraten in verschiedenen Krankheitsbereichen. |
Die "Übersetzungslücke" in der Arzneimittelentwicklung bezieht sich auf die erheblichen Schwierigkeiten und die hohe Fehlerrate, die beim Versuch auftreten, vielversprechende grundlegende wissenschaftliche Entdeckungen aus dem Labor ("Bank") in wirksame und sichere Behandlungen für menschliche Patienten ("Bett") zu übersetzen. Eine primäre Herausforderung besteht darin, dass präklinische Modelle, insbesondere Tierstudien, oft nicht genau vorhersagen können, wie sich ein potenzielles Medikament beim Menschen verhalten wird.
Darüber hinaus sind Tierversuche notorisch teuer und langsam. Schätzungen zufolge können sie die Entwicklungszeit um Monate oder sogar Jahre verlängern und die F&E-Kosten um Hunderte von Millionen Dollar erhöhen.
Tabelle: Vergleich von Kosten und Zeitplänen für traditionelle Tierversuche vs. NAMs in der präklinischen Arzneimittelentwicklung
| Aspekt | Traditionelle Tierversuche | Neue alternative Methoden (NAMs) |
|---|---|---|
| Kosten | 2–4 Millionen Dollar pro Studie; hohe Variabilität | Niedrigere Kosten aufgrund von Automatisierung und Effizienz |
| Zeitplan | 4–10 Jahre für umfassende Studien | Schnelle Ausgaben (Wochen bis Monate) |
| Effizienz | Begrenzter Vorhersagewert für die menschliche Biologie | Hochdurchsatz und menschenrelevant |
| Herausforderungen | Notwendig für komplexe, langfristige Bewertungen; regulatorische Anforderungen | Komplexe Systeme können nicht vollständig repliziert werden; begrenzte regulatorische Akzeptanz |
Dann ist da noch die ethische Berechnung. Jedes Jahr werden Zehntausende von Tieren in der US-amerikanischen Arzneimittelentwicklung eingesetzt. Viele dieser Experimente beinhalten invasive Verfahren ohne Möglichkeit der Genesung. In diesem Licht ist die FDA-Bewegung nicht nur ein wissenschaftlicher Sprung – sondern ein ethischer Wendepunkt.
Menschenrelevante Tests: Präzision ohne Schmerzen
KI-Modelle und Organoide stehen im Mittelpunkt
Was ersetzt das alte Modell? Ein leistungsstarker Zusammenfluss von Technologien, die einst als futuristisch galten:
-
Künstliche Intelligenz Toxikologie: KI-Tools, die mit Millionen von Datenpunkten trainiert wurden, können jetzt simulieren, wie sich Medikamente in menschlichem Gewebe verhalten, und Nebenwirkungen und Organtoxizität mit zunehmender Genauigkeit vorhersagen. Einige Modelle können die Verteilung monoklonaler Antikörper durch den menschlichen Körper in silico abbilden – ohne ein einziges Versuchstier.
-
Organoide und Organ-on-Chip-Systeme: Diese im Labor gezüchteten menschlichen Gewebe – winzige Versionen von Organen wie Leber, Herz oder Immunsystem – bieten einen direkten Einblick in die menschliche Biologie. Forscher können jetzt in Echtzeit beobachten, wie sich Medikamente auf tatsächliche menschliche Zellen auswirken.
Ein Erstanwender in einem mittelständischen Biotech-Unternehmen bemerkte: „In unserem jüngsten Programm für monoklonale Antikörper haben die Organoidmodelle eine Hepatotoxizität festgestellt, die unsere Rattenmodelle vollständig übersehen haben. Das ist nicht nur Effizienz. Das ist Patientensicherheit.“
Wer gewinnt, wer verliert: Eine neu ausbalancierte Branche
Gewinner: Technologiegetriebene Innovatoren und agile Biotech-Unternehmen
Für Unternehmen, die seit langem in menschenrelevante Tests investieren, ist die Entscheidung der FDA eine Rechtfertigung – und Rückenwind. Dazu gehören:
- Biotech-Unternehmen, die Organoid- und In-vitro-Systeme nutzen, in der Lage, ihre präklinischen Zeitpläne um bis zu 40 % zu reduzieren.
- KI-Plattformentwickler, die prädiktive Toxikologiemodelle entwickeln, die große Pharmaunternehmen jetzt lizenzieren wollen.
- Auftragsforschungsinstitute (CROs), die früh von Tierversuchen auf zellbasierte Assays und Siliziumsimulationen umgestellt haben.

Investoren haben dies bereits zur Kenntnis genommen. „Wir sehen dies als einen säkularen Fünfjahres-Rückenwind für jeden Akteur, der an alternativen Tests beteiligt ist“, sagte ein Portfoliomanager bei einem auf Biowissenschaften spezialisierten Hedgefonds.
Verlierer: Legacy-Tierversuchsunternehmen gefährdet
Die Auswirkungen auf traditionelle Tierversuchs-CROs sind ernüchternder. Berichte deuten auf einen Rückgang der Aktien von Charles River Laboratories um 28 % hin, einem Vorreiter für die Tierversuchsindustrie, nach der Ankündigung der FDA. Diese Firmen stehen nun vor einer klaren Wahl: sich wandeln oder verschwinden.
Aktuelle Aktienkursentwicklungstabelle für Charles River Laboratories (CRL).
| Datum | Eröffnungskurs (USD) | Höchstkurs (USD) | Tiefstkurs (USD) | Schlusskurs (USD) | Volumen |
|---|---|---|---|---|---|
| 2025-04-09 | 121.71 | 139.31 | 117.26 | 139.07 | 1,974,894 |
| 2025-04-08 | 136.86 | 137.96 | 122.06 | 123.61 | 1,586,299 |
| 2025-04-07 | 132.79 | 139.16 | 128.03 | 134.07 | 1,984,859 |
„Es ist wie der Film-zu-Digital-Übergang in der Fotografie“, sagte ein Analyst. „Nur dieses Mal geht es um Milliarden von F&E-Einnahmen.“
Der Weg nach vorn: Validierung, Übergang und Widerstand
Regulatorische Koordination und globale Auswirkungen
Während die Roadmap Klarheit bietet, ist der Übergang noch lange nicht abgeschlossen. Eine zentrale Herausforderung wird die Validierung sein – der Nachweis, dass diese neuen Methoden menschliche Ergebnisse besser vorhersagen als Tiere, und zwar konsistent und über verschiedene Arzneimitteltypen hinweg.
Validierung für New Approach Methodologies (NAMs) bezieht sich auf den Prozess der Feststellung ihrer wissenschaftlichen Zuverlässigkeit und Relevanz für einen bestimmten Verwendungszweck, wie z. B. Toxizitätstests. Dies beinhaltet typischerweise die Einhaltung definierter wissenschaftlicher Kriterien und formaler Prozesse, die oft von Aufsichtsbehörden wie der FDA geleitet werden, um sicherzustellen, dass die alternative Methode für den jeweiligen Zweck geeignet ist.
Die FDA beruft in Partnerschaft mit den NIH, dem National Toxicology Program und dem Department of Veterans Affairs eine öffentliche Werkstatt im Laufe dieses Jahres ein. Sie wird Feedback einholen und die Umsetzungszeitpläne verfeinern. Ein Pilotprogramm, das in diesem Jahr startet, wird die Roadmap unter realen Bedingungen testen und voraussichtlich breitere Leitlinien im Jahr 2026 und darüber hinaus prägen.
Andere Aufsichtsbehörden beobachten die Entwicklung genau. Sollte die FDA-Initiative erfolgreich sein, könnte eine globale Harmonisierung folgen, wobei europäische, kanadische und asiatische Aufsichtsbehörden möglicherweise dazu übergehen, Standards anzugleichen – eine Verlagerung, die die internationale Arzneimittelentwicklung erheblich rationalisieren würde.
Marktanalyse: Risiko, Rotation und Belohnung
Die Biopharma-Investmentthese im Zuge des regulatorischen Umbruchs
Aus Sicht der Kapitalmärkte ist die Bewegung der FDA eine der folgenreichsten regulatorischen Verschiebungen seit Jahrzehnten.
- Die F&E-Modelle von Biopharma werden komprimiert, wobei sich die Zeitpläne um Monate – wenn nicht sogar Jahre – verkürzen.
- Die Gewinnspannen könnten sich erweitern, da Unternehmen die Kosten reduzieren, die mit langen Tierversuchszyklen und redundanten Sicherheitsprotokollen verbunden sind.
- Die M&A-Aktivität dürfte stark ansteigen, wobei technologieaffine CROs und KI-Modellierungsfirmen zu attraktiven Zielen werden.
Prognostiziertes Marktwachstum für New Approach Methodologies (NAMs) vs. traditionelle Tierversuchsmarktprognose.
| Marktsegment | Region/Umfang | Basisjahrwert (ca.) | Prognostizierter Wert (ca.) | CAGR (ca.) | Prognosezeitraum | Quellenhinweise |
|---|---|---|---|---|---|---|
| Nicht-tierische alternative Tests (NAMs) | Global | 1,11 Milliarden USD (2019) | - | 6,34 % | 2023-2028 | Verschiedene Berichte deuten auf ein deutliches Wachstum hin, das durch ethische Bedenken und regulatorische Anstrengungen (z. B. 3R-Prinzipien) angetrieben wird. Umfasst In-vitro-, In-silico-, Organ-on-Chip-Methoden. |
| Nicht-tierische alternative Tests (NAMs) | Global | 9,8 Milliarden USD (2021) | 29,4 Milliarden USD (2030) | 13,5 % | 2022-2030 | Eine weitere Prognose, die ein noch höheres Wachstum zeigt, möglicherweise einschließlich einer breiteren Definition von NAMs oder eines anderen Marktumfangs. Betont staatliche Unterstützung und Technologien wie Organe-auf-Chip. |
| Nicht-tierische alternative Tests (NAMs) | Global | 1,8 Milliarden USD (2023) | - | 11,9 % | 2024-2032 | Hebt wachsende Investitionen, ethische Bedenken, regulatorische Unterstützung (FDA/EU) und technologische Fortschritte (Organ-on-a-Chip, 3D-Gewebemodelle) als Haupttreiber hervor. |
| In-Vitro-Toxizitätstests (Bestandteil von NAMs) | Global | 10,99 Milliarden USD (2023) | 30,06 Milliarden USD (2033) | 10,82 % | 2024-2033 | Stellt einen bedeutenden Teil des NAMs-Marktes dar und zeigt ein starkes Wachstum, das durch Arzneimittelentdeckung, chemische Sicherheit und regulatorische Akzeptanz angetrieben wird. Mehrere Quellen zeigen ähnliche CAGRs (10,8 %-11,2 %). |
| Organoid-Markt (spezifische NAM-Technologie) | Global | - | 3,3 Milliarden USD (2027) | 21,7 % | Nächste 3 Jahre | Spezifische NAM-Technologie, die ein sehr hohes Wachstumspotenzial aufweist, was die Fortschritte in der komplexen biologischen Modellierung widerspiegelt. |
| Traditionelle Tierversuche | Global | 10,74 Milliarden USD (2019) | - | 1,03 % | 2023-2028 | Zeigt ein deutlich langsameres Wachstum im Vergleich zu NAMs, was auf ethischen Druck, die Einführung von 3R-Prinzipien und regulatorische Verschiebungen (z. B. EPA-Ziel, Säugetiertests bis 2035 zu beenden) zurückzuführen ist. |
| Traditionelle Tierversuche | Global | 10,74 Milliarden USD (2019) | - | 2,64 % | 2028-2035 | Die Prognose deutet auf ein leicht erhöhtes Wachstum im späteren Zeitraum hin, das aber immer noch deutlich geringer ist als bei NAMs. |
| Tiermodellmarkt | Global | 1,9 Milliarden USD (2022) | 3,6 Milliarden USD (2032) | 6,6 % | 2023-2032 | Diese Prognose konzentriert sich speziell auf den Markt für die Tiere selbst und zeigt ein moderates Wachstum, das wahrscheinlich durch den laufenden F&E-Bedarf trotz des Drängens nach Alternativen angetrieben wird. |
Gleichzeitig lauert das Risiko für langsame Anwender. „Investoren müssen äußerst selektiv sein“, sagte ein Fondsmanager. „Dies ist keine steigende Flut. Es ist eine Flutwelle. Einige Boote werden aufsteigen. Andere werden sinken.“
Eine stille, humane Revolution
Die Entscheidung der FDA, obligatorische Tierversuche für monoklonale Antikörper zu beenden, ist mehr als nur ein regulatorisches Update. Es ist der stille Beginn einer biomedizinischen Transformation, die schnellere Heilungen, niedrigere Medikamentenpreise, bessere Wissenschaft und weniger Käfige verspricht.
Und für die anspruchsvollsten Investoren der Welt markiert sie den Beginn einer neuen Investitionsära – einer Ära, die technologische Weitsicht, ethische Führung und eine am Menschen orientierte Wissenschaft belohnt.
In einer Welt des Lärms beginnen die bedeutungsvollsten Revolutionen manchmal mit dem leisen Summen eines Siliziumchips – der das Quietschen einer Laborratte ersetzt.